|
Powersuche
| |
Oxidation
Übersicht über die verschiedenen Oxidationsstufen
| Alkohol |
Aldehyd |
Carbonsäure |
| Alkan |
Keton |
|
zurück zum Inhaltsverzeichnis
Will man nun vom Alkohol zum Aldehyd bzw. Keton oxidieren, so gestaltet es sich schon mal als relativ schwierig, auf dieser Stufe stehen zu bleiben, spricht nicht das Produkt weiter bis zur Carbonsäure zu oxidieren. Des weiteren können sich im Molekül verschiedene Hydroxy-Gruppen befinden, von denen man aber nur eine bestimmte zum Aldehyd/Keton oxidieren möchte (Selektivität).
nach einem Vortrag von Jan Michael Klitschke
Bei der Bayer-Villiger Oxidation werden säurekatalysiert Ketone zu Carbonsäureestern (bzw. Lactonen) oxidiert. Dies entspricht formal einer Insertion eines Sauerstoffatoms in eine der C-C Bindungen an der Carbonylgruppe.
Standartreagenzien sind: 3-Chlorperoxybenzoesäure (MCPBA metachlorperoxybenzoicacid und Triflourperoxyessigsäure (mit Na2HPO4-Puffer)
Die Insertion des Sauerstoffatoms erfolgt durch Addition der Peroxycarbonsäure an die Carbonylgruppe und Wanderung eines Restes an den Sauerstoff, wie der Mechanismus nach Criegee [Lit. 2] am Beispiel mit MCPBA zeigt.
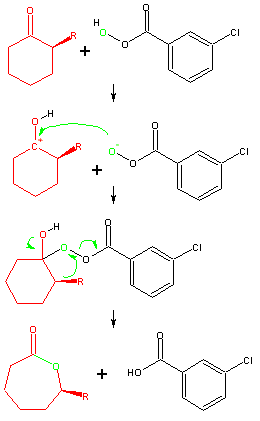
Wird in der Reaktion ein unsymmetrisches Keton eingesetzt, so hängt die Struktur des Produkts davon ab, welche der beiden Gruppen wandert. Aus kinetischen Studien [Lit. 3] hat sich folgende Wanderungstendenz ergeben:
tert. Alkyl > sek. Alkyl > prim. Alkyl und tert. Alkyl > sek. Alkyl > Benzyl und Phenyl > prim. Alkyl > Methyl
Die Wanderung des Restes erfolgt unter Retention der Konfiguration.
Enzymatische Bayer-Villiger-Oxidationen erlauben z.B. die hoch enantioselektive Synthese chiraler Lactone aus achiralen Ketonen (meso-Cyclohexanonen) [Lit. 4-6].
Mit Bis(trimethylsilylperoxid) können auch ungesättigte Ketone oxidiert werden [Lit. 7]. zumal die gängigen Standartreagenzien, wie Trifluorperoxyessigsäure, hier sonst die C=C - Doppelbindung angreifen würden.
Literatur
[1] March, S 1098f
[2] R. Crigee, Ann., 560, 127 (1948)
[3] W.D. Emmons, J. Am. Chem. Soc., 1958, 80, S 6393ff
[4] M.J. Taschner, D.J. Black, J. Am. Chem. Soc., 1988, 110, S 6892f
[5] C.T. Walsh, J. Chem, Angew. Chem., 1988, 100, S 342ff
[6] M. M. Kayser, J. Chen, J. Org. Chem., 1998, 63, S 7103ff
[7] M. Suzuki, H. Takada, r. Noyori, J. Org. Chem., 1982, 47, S 902f
nach einem Vortrag von Matthias Elhardt
Geeignet für prim. Alkohol --> Aldehyd und sek. Alkohol --> Keton
Vorteile der Swern-Oxidation
- Selektive Oxidation von prim. Alkohol zum Aldehyd bzw. sek. Alkohol zum Keton
- Mittels Swern Oxidation kann eine ganze Bandbreite von Alkoholen oxidiert werden, ohne überoxidiert zu werden.
- Keine Isomerisierungseffekte bei Doppelbindungen
- i.a. hohe Ausbeuten bei geeigneter Wahl der Reaktionsparameter (Temperatur, Base, etc.)Reaktion verläuft i.A. schnell
- Aus Oxalylchlorid entstehen Gase (CO2,CO)
- Im Vergleich zu Chromreagenzien harmlose Nebenprodukte
Nachteile
- Tiefe Reaktionstemperatur (sonst Zerstörung des aktiven DMSOs)
- Funktioniert mit den meisten Allenalkoholen nicht.
- Bei Heteroaromaten oder Heterocyclen kann das Heteroatom auch oxidiert werden
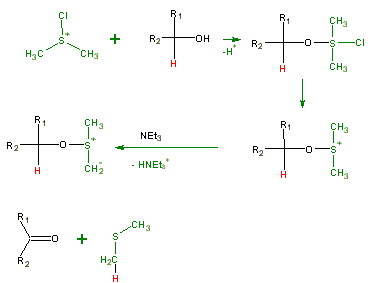
Herstellung des aktivierten DMSO
Die Herstellung des für die Reaktion benötigten aktivierten DMSO erfolgt meistens unmittelbar vor der eigentlichen Oxidation. Man geht hierbei von Dimethylsulfoxid (DMSO) und Oxalylchlorid (Ethandisäuredichlorid) aus. Die Übertragung eines Chlorid-Ions führt zum sogenannten aktivierten DMSO, das das eigentliche Oxidationsmittel darstellt. Die Reaktion wird bei 60°C bis 20°C in Dichlormethan durchgeführt.

Die eigentliche Oxidation eines primären oder sekundären Alkohols erfolgt dann mit Hilfe der Base Triethylamin (NEt3), die ein H+ Ion von einer der Methylgruppen des DMSO abstrahiert. Daraufhin abstrahiert diese Methylengruppe das Wasserstoffion des Alkohols und Dimethylsulfid wird abgespalten.
Literatur
K. Omura und D. Swern, Tetrahedron 1978, Vol. 34, 1651-1660
A. Mancuso, D. Brownfain, D. Swern, J. Org. Chem., 1979, Vol. 44, No. 23
A. Mancuso, S.-L. Huang, D. Swern, J. Org. Chem., 1978, Vol. 43, No. 12
A. Mancuso und D. Swern, Synthesis, 1981, S 165
R. Brückner, Reaktionsmechanismen, Spektrum Akademischer Verlag, 1996
nach einem Vortrag von Heiner Meister
Die Oxidationswirkung von Cr(VI) ist gut durch Reaktionsbedingungen steuerbar, was eine hohe Selektivität durch geeignete Wahl der Bedingungen ermöglicht. Einen Einfluss haben:
pH-Wert: H+ katalysiert und erhöht somit die Oxidationswirkung im Sauren
Lösungsmittel: e--Donatoren bilden mit Chromat Komplexe (z.B. Pyridin, HMPT, DMSO) und erniedrigen somit die Oxidationswirkung (milder).
Substituenteneinfluß: mit Nucleophilen (Cl-, AcOAC, HO-tBu) bildet Cr(VI) O2CrCl2, AcO-CrO2-OAc und tBuO-CrO2-OtBu, welche in organischen Lösungsmitteln löslich sind und wasserfreies, neutrales und somit mildes arbeiten ermöglichen.
Ausgewählte Reagenzien
Jones Reagenz
Wäßrig, verd. H2SO4 mit CrO3 gesättigt + Aceton im Überschuss (schützt Produkt vor weiterer Oxidation)
Ermöglicht die Oxidation von prim. Alkoholen zu Carbonsäuren und von sekundären Alkoholen zu Ketonen.
Collin Reagenz
Wasserfrei (milder), CrO3 + Pyridin (in H2CCl2)
Oxidation von prim. Alkoholen zu Aldehyden und sek. Alkoholen zu Ketonen.
Die Oxidation von Alkanen ist zumeist schwierig steuerbar, es bildet sich eine Vielzahl an Produkten.
Literatur:
G. Cainelli, G. Cardillo, Chromium Oxidations in org. Chemistry, Springer Verlag 1984
R. Bruckner, Reaktionsmechanismen, Spektrum Verlag
Fuhrhop, Penzlin, Organic Synthesis, VCH
nach einem Vortrag von Bernd Neumann
Eine relativ neue Methode gestattet die Oxidation von primären und sekundären Alkoholen zu den jeweiligen Aldehyden bzw. Ketonen, wobei Sulfide, Enolether , Epoxide, Furane und sekundäre Amine nicht oxidiert werden.
Des weiteren werden benzylische Alkohole gegenüber aliphatischen Alkoholen selektiv oxidiert.
Als Nachteil erweist sich, dass aktivierte Amine z.T. auch oxidiert werden (z.B.N-Benzylbenzamid zu Benzaldehyd).
Zur Oxidation werden organischen Iod-(V)-Reagenzien verwendet, meist DMP (Darstellung nach [Lit 3]).
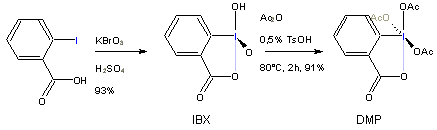
Es gibt abgewandelte Methoden mit IBX (in DMSO) als Oxidationsmittel, oder mit weiteren ähnlichen Iod-(V)-Reagenzien.
IBX ist explosionsgefährlich bei über 200°C oder Stoß; DMP ist auch käuflich, zersetzt sich allerdings allmählich.
Reaktionsbedingungen
- Raumtemperatur, vgl. Swern-Oxidation, neutrale oder leicht saure Lösung in CHCl3, CH2Cl2, CH3CN
- Kurze Reaktionsdauer (typisch: 0,5 2h); Inertatmosphäre wird nicht unbedingt benötigt
- Kein hoher Überschuss an Oxidationsmittel benötigt (typisch 5-10%)
- Einfache Aufarbeitung (Hydrolyse der entstehenden Iod-(I)-Spezies mit verd. NaOH, Destillation des Produkts), vgl. Cr-(VI)-Reagenzien
- i.A. hohe Ausbeuten
- DMP muss nicht direkt vor der Reaktion hergestellt werden, vgl. Swern-Oxidation
Die Zugabe von Alkoholen , auch des nicht oxidierbaren tBuOH, sowie von Wasser wirkt oft reaktionsbeschleunigend , allerdings wird dann bei Alkoholen die Hydrolyse oft erschwert [Lit: 5].
Literatur:
[1] Dess,Martin; J. Org. Chem., 1983, 48, 4156-4158
[2] Dess, Martin; J. Am. Chem. Soc., 1991, 113, 7277-7287
[3] Ireland, Liu; J. Org. Chem., 1993, 58, 2899
[4] Munari; J. Org. Chem., 1996, 61, 9272-9279
[5] Meyer, Schreiber; J. Org. Chem. 1994, 59, 7549-7552
zurück zum Inhaltsverzeichnis
|
|
